
Introduction
Technical documentation, also known as technical file, is a set of the documents that contains all the technical information concerning a medical device. Therefore, every EU manufacturer of medical devices (regardless of risk class) must according to regulations create technical documentation. This must include clear information regarding design, manufacture, and operation of the device.
The journey manufacturers need go through to meet regulatory requirements – and get the CE mark – is often very complex. An example of this process is presented in Figure 1 below.
In this article, we will focus on the MDR requirements. The MDR (Medical Device Regulation 2017/745) replaces MDD (Medical Device Directive 93/42/ECC). For MDD devices already on the market the transition to MDR is often described as “demanding” and “expensive”. However, it is good to keep in mind that by creating a well-planned regulatory strategy and preparing the technical documentation “as good as possible from the beginning” can facilitate a smooth process.
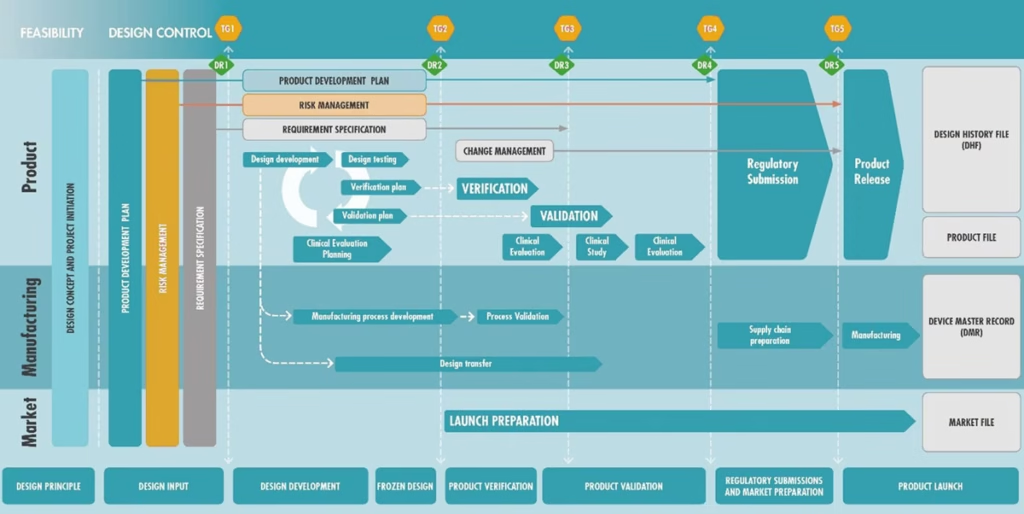
Figure 1. The process of the device launching
What are the obligations of the manufacturer?
A manufacturer of the medical device is obligated to:
- prepare technical documentation before placing a device on the market;
- supply technical documentation on devices greater than class I, for review and approval of a Notified Body before making the device available on the market;
- maintain the technical documentation throughout the device’s entire life cycle;
- file technical documentation available for Competent Authorities (CAs) for 10 years after previous device was placed on the market. Or 15 years for implantable devices.
The technical documentation must show that the medical device conforms to applicable regulatory requirements such as MDR. Which therefore ensures that the medical device is safe and effective.
Key components of technical documentation
Key components of the technical documentation are specified in Annex II and Annex III in the MDR and “shall be presented in a clear, organized, readily searchable and unambiguous manner” (Annex II, point 1.1.1 (a)):
Annex II – Technical Documentation
- Device Description and Specification, including variants and accessories.
- Device Description and Specification
- Reference to previous and similar generations of the device
- Information to be supplied by the manufacturer
- Design and Manufacturing Information
- General Safety and Performance Requirements
- Benefit-Risk Analysis and Risk Management
- Device Verification and Validation
- Pre-clinical and Clinical data
- Additional information required in specific cases.
Annex III – Technical Documentation on Post Market Surveillance
- The Post Market Surveillance Plan
- The PSUR (Periodic Safety Update Report)
- PMS Report
How to create and maintain the technical documentation?
As mentioned above, the technical documentation preferably presents as clear, organised, and unambiguous. Also, following applicable standards and guidelines increases the probability of good technical documentation which is easy to maintain. The Notified Body, which the manufacturer has a signed contract with, usually prepares a technical guidance with clear instruction on how the technical documentation should be submitted. To follow this guidance can increase the speed of the Notified Body review. The support of an experienced person, for instance a consultant, can enable this process. Since the technical documentation is often considerable, the choice of storage is important. Using an electronic signing and storage solution can save a lot of time. It can also improve the possibility of maintaining the technical documentation up to date during the device’s entire life cycle.
Common challenges and solutions
A common challenge is estimating the time and resources needed for a Notified Body submission. Project planning, as well as having available resources to answer the Notified Bodies questions, play a crucial role to get approval and getting the device on the market. The support of an experienced person, for instance a consultant can enable a smooth process.
Conclusion
As presented, the preparation of technical documentation is a requirement that needs to be fulfilled to place a device on the European market. Once it is done, it is important to keep the technical documentation up to date according to the latest regulations. Compared with the MDD requirements, the expected quality of the technical documentation has increased in conjunction with the MDR transition. The inclusion of the PMS requirements, according to Annex III of the MDR and stricter requirements on clinical data, mean that manufacturers must improve the scientific quality of technical documentation. At Key2Complaince, we will help you every step of the way when creating a technical documentation, and also keeping it up to date.